BIOMOL is a research project at Dowling College to augment Rasmol's [3] [7] existing macromolecular display functions with new capabilities by taking advantage of recent increases in local computing power in order to move functionality that is now scattered among various local and remote systems into one local package. Most entries determined by x-ray diffraction provided by the Protein Data Bank [2] [1] contain the crystallographic asymmetric unit, or unique fragments in the case of molecules such as viruses. In order to understand the biological significance of this data, it is important to apply both the crystallographic symmetry and appropriate non-crystallographic symmetry operations. EBI offers biological units in the PQS database [5]. (See pqs.ebi.ac.uk/pqs-doc/pqs-doc.shtml). The RCSB offers biological unit images and coordinates for many structures, further responding to this important need (see beta.rcsb.org/pdb/beta.html). However, the use of a shared server combined with the speed limitations of remote internet access limits the volume of data that can be efficiently provided. With appropriate extensions to the tools available to users on their local machines, many more choices can be made available in a cost-effective manner, allowing more timely insights by researchers and more effective training of students. By bringing full management of crystallographic and non-crystallographic symmetry operations into RasMol a better understanding of biological units is possible without the user having to retrieve information from multiple sources. Having symmetry and biological unit generation within RasMol also allows work with coordinate entries that have not yet been deposited in the PDB. In addition, when working intensively with a particular structure and needing to look at other entries, it is clearly desirable to provide local versions of search tools which interact smoothly and naturally with the display functions. We are in the last year of this three year project to extend RasMol to increase local functionality in management of biological units and to create real-time integration with both the existing web-based search engines and with local versions to achieve higher performance.
Near the end of the second year of the project, the grant was supplemented with an award to create a new Wide Protein Data Bank Format (WPDB). The Protein Data Bank is the world-wide repository for the results of macromolecular structure determination. The macromolecular CIF (mmCIF) format [4] was developed to capture all the detailed searchable relationships among data items and to overcome the field-width limitations of the old 80-column PDB format [2][6]. While mmCIF has been of great value to the internal operation of the PDB and has simplified the creation of powerful search engines, users of the PDB have been reluctant to make the transition from the easily-read fixed-field PDB format to mmCIF, and most existing macromolecular software remains unable to read mmCIF directly. We are addressing the issue by creating a new fixed-field wide PDB format (WPDB) that carries all the information provided by mmCIF using record formats very similar to those in the existing PDB format. By increasing the widths of many fields (especially atom number, atom name, chain identifier and coordinates), we are overcoming the major deficiencies of the old PDB format, including the 99,999 atom limitation and the idiosyncratic atom naming forced by the 4-column field width, especially for hydrogens. In addition, we are adding new record types to carry the new information from the mmCIF files (e.g. the more detailed information on secondary structure and biological function). We are creating simple external translation filter programs to go back and forth to mmCIF format and, where the data permits, to the existing PDB format. Software developers should find it completely straightforward to revise their input and output routines to process wide PDB (WPDB) and anyone will be able to use the filters.
PI/PD: Herbert J. Bernstein (Dowling College)
Research Collaborator: Frances C. Bernstein (Bernstein + Sons)
Current Students: Isaac Awuah Asiamah, Niroshan Egodawatte
Students who worked on the project in prior years: Sagar Pilania, Vencislav Stanev, Clarice Chigbo, Ricky Chachra, Georgi Darakev, Nicolay Darakev, Parag Jain, Stavros Louris, Georgi Todorov
In the two year and three quarters of the project, coding was started on almost all aspects of the project, and most of the major components are working. The essential crystallographic symmetry code has been working since the first summer of the project. A first version of the non-cystallographic symmetry (NCS) code was also in place early on. It is now being reworked to allow better memory management when working with very large numbers of NCS operations. The connected component code needed to infer biological activity is working. The WPDB format has been integrated with RasMol for both reading and writing.
The major open items are software integration and documentation, which seem achievable within the remaining 3.5 months of the project.
The work done to date and comments from users of RasMol have suggested extension of the project to cover heterogeneous complexes and to make more extensive use of the new WPDB format to address the issues of generated structures of more than 99,999 atoms, for which further funding has been requested.
The project web site, http://biomol.arcib.org, now includes a sub-site for WPDB,http://biomol.arcib.org/WPDB/.
As reported earlier, some of the code needed for this project to efficiently identify neighboring atoms proved useful for the work we are doing on an NSF funded project on molecular surfaces (and vice versa), so this DOE grant has been credited on the RasMol 2.7.3 web page (http://www.bernstein-plus-sons.com/software/RasMol_2.7.3) along with NSF grants DBI-0203064, DBI-0315281 and EF-0312612. The RS/6000s that were transferred from BNL have been integrated into the laboratory network and have been used as intended to do cross-platform validation of RasMol. The problems with sensitivity of this old equipment to high temperatures reported earlier seem now to be under control, but we can only be sure when we reach higher summer temperatures.
The initial effort in the project was the integration of code for processing of crystallographic symmetry with the
code for RasMol. This work was started by Vencislav Stavev and continued by Sagar Pilania and Stavros Louris. This
resulted in a patch to rasmol (see http://biomol.arcib.org/BIOMOL/symm0/symm0.patch)
that can accept symmetry from a dictionary file (see the short example
at http://biomol.arcib.org/BIOMOL/symm0/symop.dic).
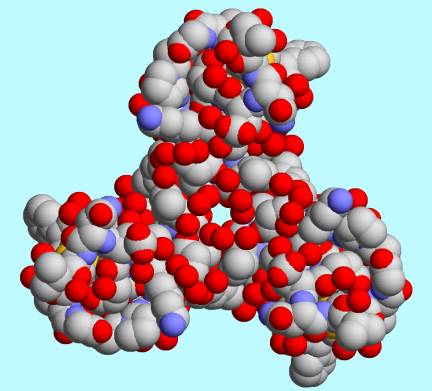
This code was used by Sagar Pilania to generate
an interesting image with a modified version of RasMol.
Starting with the asymmetric unit of the PDB entry 1AJJ [8] [9] shown at
http://biomol.arcib.org/BIOMOL/symm0/1ajj.jpg he applied a three-fold symmetry operation and drew the lovely picture
shown at http://biomol.arcib.org/BIOMOL/symm0/1ajj_threefold.jpg
.
The next step was to reformat the dictionary and expand it to a full list of symmetry operations for all space
groups in all common settings. There are three major possible choices in making such a list available to a
program: use of a program to generate symmetry operations from space group symbols on the fly, use of a web-server
to do the translation, and use of a complete pre-compiled table. We have chosen the last option. The first
approach is, arguably, the most efficient one in terms of program memory demands and time. However, one of the
goals of this project is to make all the fruits of its efforts available as open-source code. Use of one of the
existing programs within the context of an open-source license such as the GPL requires will require c
areful negotiations with the authors. Creation of yet another such program would be reinvention of a well-designed
wheel. Use of a remote web site interactively would conflict with the performance goals of the project. Therefore
we have chosen to ensure the stability and distributability of a base level of the code by providing a
complete pre-compiled table. Arrangement for embedded use of one of the existing programs will be pursued later
(see below). This work was done by Stavros Louris with checking by Isaac Awuah Asiamah and resulted in the updated
dictionary at http://biomol.arcib.org/BIOMOL/symm1/symop.dic. The initial effort to produce this dictionary was
based on manual references against Bernhard Rupp's Crystallography 101 web site at
http://www-structure.llnl.gov/Xray/101
index.html
http://www-structure.llnl.gov/X
ray/tutorial/spcgrps.htm
using the program SEXIE [10]. The results were then
validated against Volume A of the International Tables of Crystallography
[11]. The error rate from this manual approach was
too high and therefore the table was redone by fully automated
use of Ralf Grosse-Kunstleve's program sginfo
[12] and a set of simple scripts.
Grosse-Kunstleve has graciously provided permission to incorporate appropriate portions of sginfo within RasMol
under its open-source license, but further negotiations will be needed once the code is completed if the
arrangement is to be formalized. Until and unless such an arrangement is made, the BIOMOL project is using only
one list of space group identifications in a script with small supporting programs to generate the necessary
table. See the script and cpp files in
http://biomol.arcib.org/BIOMOL/symm1/.
The current dictionary is
covered by the open-source license and is sufficient for the immediate needs of the project.
he applied a three-fold symmetry operation and drew the lovely picture
shown at http://biomol.arcib.org/BIOMOL/symm0/1ajj_threefold.jpg
.
The next step was to reformat the dictionary and expand it to a full list of symmetry operations for all space
groups in all common settings. There are three major possible choices in making such a list available to a
program: use of a program to generate symmetry operations from space group symbols on the fly, use of a web-server
to do the translation, and use of a complete pre-compiled table. We have chosen the last option. The first
approach is, arguably, the most efficient one in terms of program memory demands and time. However, one of the
goals of this project is to make all the fruits of its efforts available as open-source code. Use of one of the
existing programs within the context of an open-source license such as the GPL requires will require c
areful negotiations with the authors. Creation of yet another such program would be reinvention of a well-designed
wheel. Use of a remote web site interactively would conflict with the performance goals of the project. Therefore
we have chosen to ensure the stability and distributability of a base level of the code by providing a
complete pre-compiled table. Arrangement for embedded use of one of the existing programs will be pursued later
(see below). This work was done by Stavros Louris with checking by Isaac Awuah Asiamah and resulted in the updated
dictionary at http://biomol.arcib.org/BIOMOL/symm1/symop.dic. The initial effort to produce this dictionary was
based on manual references against Bernhard Rupp's Crystallography 101 web site at
http://www-structure.llnl.gov/Xray/101
index.html
http://www-structure.llnl.gov/X
ray/tutorial/spcgrps.htm
using the program SEXIE [10]. The results were then
validated against Volume A of the International Tables of Crystallography
[11]. The error rate from this manual approach was
too high and therefore the table was redone by fully automated
use of Ralf Grosse-Kunstleve's program sginfo
[12] and a set of simple scripts.
Grosse-Kunstleve has graciously provided permission to incorporate appropriate portions of sginfo within RasMol
under its open-source license, but further negotiations will be needed once the code is completed if the
arrangement is to be formalized. Until and unless such an arrangement is made, the BIOMOL project is using only
one list of space group identifications in a script with small supporting programs to generate the necessary
table. See the script and cpp files in
http://biomol.arcib.org/BIOMOL/symm1/.
The current dictionary is
covered by the open-source license and is sufficient for the immediate needs of the project.
The next phase of the project was a rework of the initial symmetry generation code to make use of the full dictionary and to integrate it with the most current release of RasMol, release 2.7.3. The code for the work done on this phase to 24 May 2005 is available as a patch for RasMol 2.7.3 (see http://biomol.arcib.org/BIOMOL/symm1/symm1.patch).
The major changes since then have been to restructure the code to allow for many more atoms by replacing forward pointers for symmetry operations with back pointers for each generated atom to the root. Uses of small-grained dynamic memory allocation have been sharply reduced. Most of the short space group names used in the PDB have been mapped to their full names, making use of cell parameters to disambiguate the handling of H3 and R3.
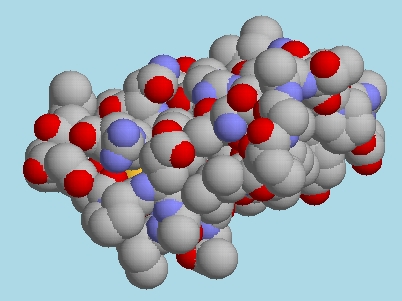
The new code is being tested, but is already producing some
interesting images, such as the expansion of the two-fold P 21
symmetry in 1CRN [13]. The
asymmetric unit can be seen in the image at
http://biomol.arcib.org/BIOMOL/symm1/1crn.jpg
 and the expanded twofold can be seen
in the image at
http://biomol.arcib.org/BIOMOL/symm1/1crn_twofold.jpg
and the expanded twofold can be seen
in the image at
http://biomol.arcib.org/BIOMOL/symm1/1crn_twofold.jpg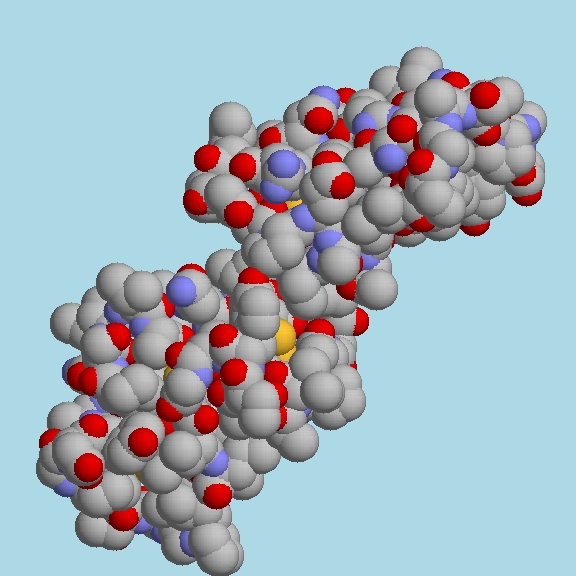 .
.
In the third phase of the project we are bringing together the work done in the prior phases and the new work on
WPDB to make one integrated system. WPDB read and write capabilities have been added to RasMol. The current best
patch for RasMol to enable WPDB functionality to date is at
http://biomol.arcib.org/WPDB/wpdb1.patch.
For crystallographic symmetry, the table of space groups is well populated, but a few omissions of alternate
settings have been discovered in testing that will have to be inserted. An earlier limitation on the number of
non-crystallographic symmetry operations has
been corrected. Crystallographic and non-crystallographic symmetry can be used together as in the image from PDB
entry 1LDB shown with both non-crystallographic and then crystallographic symmetry expanded at
http://biomol.arcib.org/BIOMOL/symm2/1ldb_expanded.jpg . The best current cumulative code patches for the crystallographic and non-crystallographic symmetry calculations are in
http://biomol.arcib.org/BIOMOL/symm2/symm2.patch.
We also note that movie-making scripts have been provided in http://biomol.arcib.org/BIOMOL/wmovies.
. The best current cumulative code patches for the crystallographic and non-crystallographic symmetry calculations are in
http://biomol.arcib.org/BIOMOL/symm2/symm2.patch.
We also note that movie-making scripts have been provided in http://biomol.arcib.org/BIOMOL/wmovies.
[1] H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. The Protein Data Bank. Nucleic Acids Research, 28:235-242, 2000. See http://www.rcsb.org.
[2] F. C. Bernstein, T. F. Koetzle, G. J. B. Williams, E. F. Meyer Jr., M. D. Brice, J. R. Rodgers, O. Kennard, T. Shimanouchi, and M. Tasumi. The protein data bank: a computer-based archival file for macromolecular structures. J. Mol. B iol., 112:535-542, 1977.
[3] H. J. Bernstein. Recent changes to RasMol, recombining the variants. Trends in Biological Sciences (TIBS), 25(9):453-455, September 2000.
[4] P. E. Bourne, H. M. Berman, B. McMahon, K. D.. Watenpaugh, J. Westbrook, and P. M. D. Fitzgerald. The macromolecular crystallographic information file (mmCIF). Methods in Enzymology, 277:571-590, 1997.
[5] K. Henrick and J. Thornton. PQS: A protein quaternary structure file server. Trends in Biochemical Sciences (TIBS), 9:358-361, 1998.
[6] Protein Data Bank Atomic Coordinate and Bibliographic Entry Format Description. Technical report, Brookhaven National Laboratory, 1992. http://arcib.dowling.edu/÷BernsteH/PDB format 1992.pdf.
[7] Roger Sayle and E. James Milner-White. RasMol: Biomolecular graphics for all. Trends in Biochemical Sciences (TIBS), 20(9):374, September 1995.
[8] D. Fass, S. Blacklow, P. S. Kim and J. M. Berger, PDB entry 1AJJ, LDL Receptor Ligand-Binding
Module 5, Calcium-Coordinating, 4 May 1997.
[9] D. Fass, S. Blacklow, P. S. Kim and J. M. Berger, ""Molecular basis of familial
hypercholesterolaemia from structure of LDL receptor module"" Nature 388 pp. 691, 1997.
[10] B. Rupp, B. Smith and J. Wong, "SEXIE - a Microcomputer Program for the Calculation of
Coordination Shells and Geometries", Comp. Phys. Commun. 67, 543 1992. Available on-line via
www-structure.llnl.gov/xray/tutorial/spcgrps.htm.
[11] T. Hahn, ed., "International Tables for Crystallography Volume A: Space-group symmetry",
Dordrecht: Kluwer Academic Publishers, 2002.
[12] R. W. Grosse-Kunstleve, "SgInfo - A comprehensive Collection of ANSI C Routines for the
Handling of Space Group Symmetry", 1995.www.kristall.ethz.ch/LFK/software/sginfo/
[13] W. A. Hendrickson and M. M. Teeter, "PDB entry 1CRN, Plant Seed Protein", 31 April
1981.